

Translational Activation of ATF4 through Mitochondrial Anaplerotic Metabolic Pathways Is Required for DLBCL Growth and Survival
Diffuse large B-cell lymphomas (DLBCL) are broadly dependent on anaplerotic metabolism regulated by mitochondrial SIRT3. Herein we find that translational upregulation of ATF4 is coupled with anaplerotic metabolism in DLBCLs due to nutrient deprivation caused by SIRT3 driving rapid flux of glutamine into the tricarboxylic acid (TCA) cycle. SIRT3 depletion led to ATF4 downregulation and cell death, which was rescued by ectopic ATF4 expression. Mechanistically, ATF4 translation is inhibited in SIRT3-deficient cells due to the increased pools of amino acids derived from compensatory autophagy and decreased glutamine consumption by the TCA cycle. Absence of ATF4 further aggravates this state through downregulation of its target genes, including genes for amino acid biosynthesis and import. Collectively, we identify a SIRT3-ATF4 axis required to maintain survival of DLBCL cells by enabling them to optimize amino acid uptake and utilization. Targeting ATF4 translation can potentiate the cytotoxic effect of SIRT3 inhibitor to DLBCL cells. SIGNIFICANCE: We discovered the link between SIRT3 and ATF4 in DLBCL cells, which connected lymphoma amino acid metabolism with ATF4 translation via metabolic stress signals. SIRT3-ATF4 axis is required in DLBCL cells regardless of subtype, which indicates a common metabolic vulnerability in DLBCLs and can serve as a therapeutic target.
Journal: Blood Cancer Discovery PMID: 35019856 DOI: 10.1158/2643-3230.BCD-20-0183
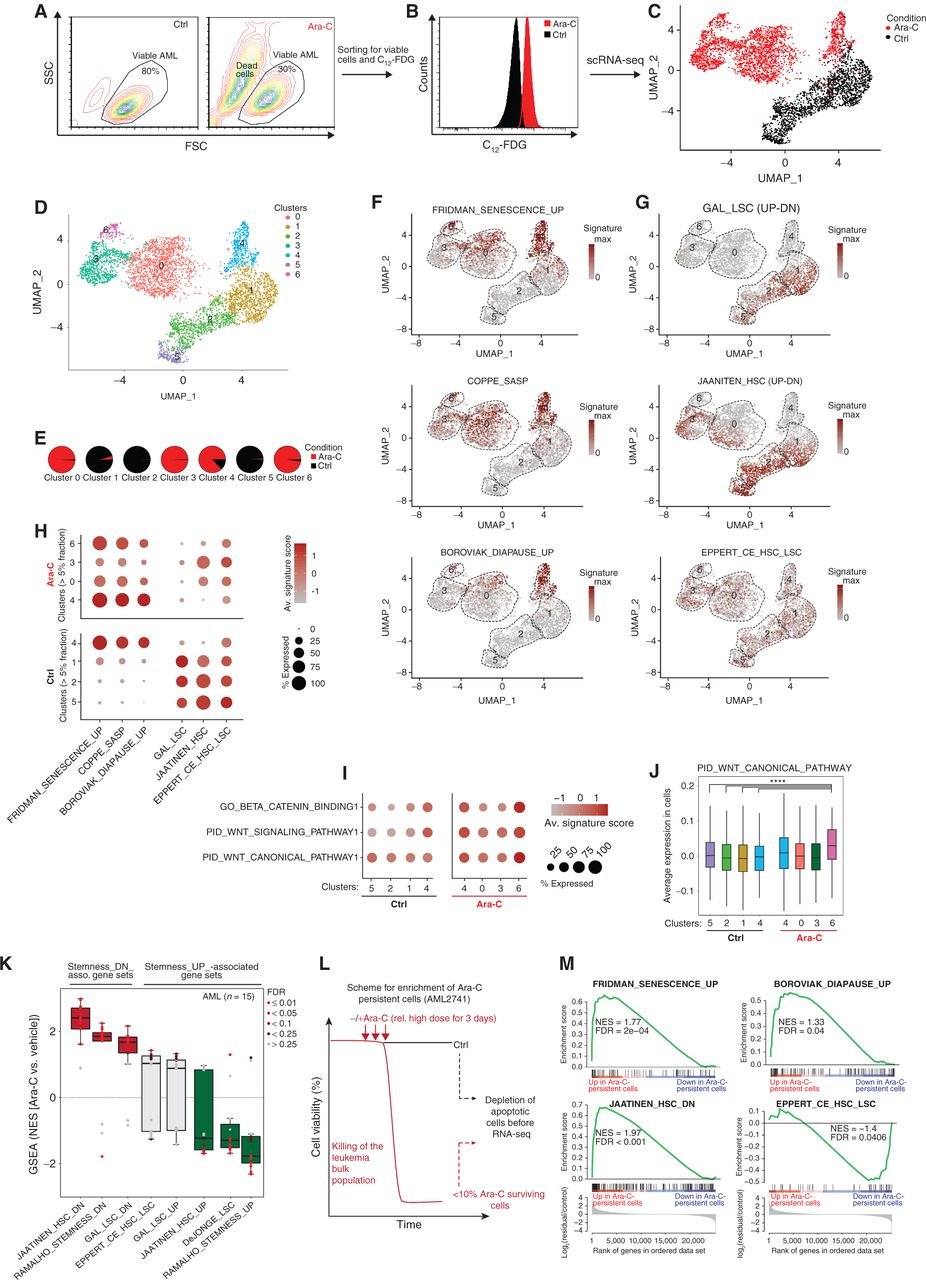
Chemotherapy Induces Senescence-Like Resilient Cells Capable of Initiating AML Recurrence
Patients with acute myeloid leukemia (AML) frequently relapse after chemotherapy, yet the mechanism by which AML reemerges is not fully understood. Herein, we show that primary AML cells enter a senescence-like phenotype following chemotherapy in vitro and in vivo. This is accompanied by induction of senescence/inflammatory and embryonic diapause transcriptional programs, with downregulation of MYC and leukemia stem cell genes. Single-cell RNA sequencing suggested depletion of leukemia stem cells in vitro and in vivo, and enrichment for subpopulations with distinct senescence-like cells. This senescence effect was transient and conferred superior colony-forming and engraftment potential. Entry into this senescence-like phenotype was dependent on ATR, and persistence of AML cells was severely impaired by ATR inhibitors. Altogether, we propose that AML relapse is facilitated by a senescence-like resilience phenotype that occurs regardless of their stem cell status. Upon recovery, these post-senescence AML cells give rise to relapsed AMLs with increased stem cell potential. SIGNIFICANCE: Despite entering complete remission after chemotherapy, relapse occurs in many patients with AML. Thus, there is an urgent need to understand the relapse mechanism in AML and the development of targeted treatments to improve outcome. Here, we identified a senescence-like resilience phenotype through which AML cells can survive and repopulate leukemia.
Journal: Cancer Discovery PMID: 33500244 DOI: 10.1158/2159-8290.CD-20-1375

Non-oncogene Addiction to SIRT3 Plays a Critical Role in Lymphomagenesis
Diffuse large B cell lymphomas (DLBCLs) are genetically heterogeneous and highly proliferative neoplasms derived from germinal center (GC) B cells. Here, we show that DLBCLs are dependent on mitochondrial lysine deacetylase SIRT3 for proliferation, survival, self-renewal, and tumor growth in vivo regardless of disease subtype and genetics. SIRT3 knockout attenuated B cell lymphomagenesis in VavP-Bcl2 mice without affecting normal GC formation. Mechanistically, SIRT3 depletion impaired glutamine flux to the TCA cycle via glutamate dehydrogenase and reduction in acetyl-CoA pools, which in turn induce autophagy and cell death. We developed a mitochondrial-targeted class I sirtuin inhibitor, YC8-02, which phenocopied the effects of SIRT3 depletion and killed DLBCL cells. SIRT3 is thus a metabolic non-oncogene addiction and therapeutic target for DLBCLs.
Journal: Cancer Cell PMID: 31185214 DOI: 10.1016/j.ccell.2019.05.002

Rational Targeting of Cooperating Layers of the Epigenome Yields Enhanced Therapeutic Efficacy against AML
Disruption of epigenetic regulation is a hallmark of acute myeloid leukemia (AML), but epigenetic therapy is complicated by the complexity of the epigenome. Herein, we developed a long-term primary AML ex vivo platform to determine whether targeting different epigenetic layers with 5-azacytidine and LSD1 inhibitors would yield improved efficacy. This combination was most effective in TET2mut AML, where it extinguished leukemia stem cells and particularly induced genes with both LSD1-bound enhancers and cytosine-methylated promoters. Functional studies indicated that derepression of genes such as GATA2 contributes to drug efficacy. Mechanistically, combination therapy increased enhancer–promoter looping and chromatin-activating marks at the GATA2 locus. CRISPRi of the LSD1-bound enhancer in patient-derived TET2mut AML was associated with dampening of therapeutic GATA2 induction. TET2 knockdown in human hematopoietic stem/progenitor cells induced loss of enhancer 5-hydroxymethylation and facilitated LSD1-mediated enhancer inactivation. Our data provide a basis for rational targeting of cooperating aberrant promoter and enhancer epigenetic marks driven by mutant epigenetic modifiers.
Journal: Cancer Discovery PMID: 31076479 DOI: 10.1158/2159-8290.CD-19-0106