
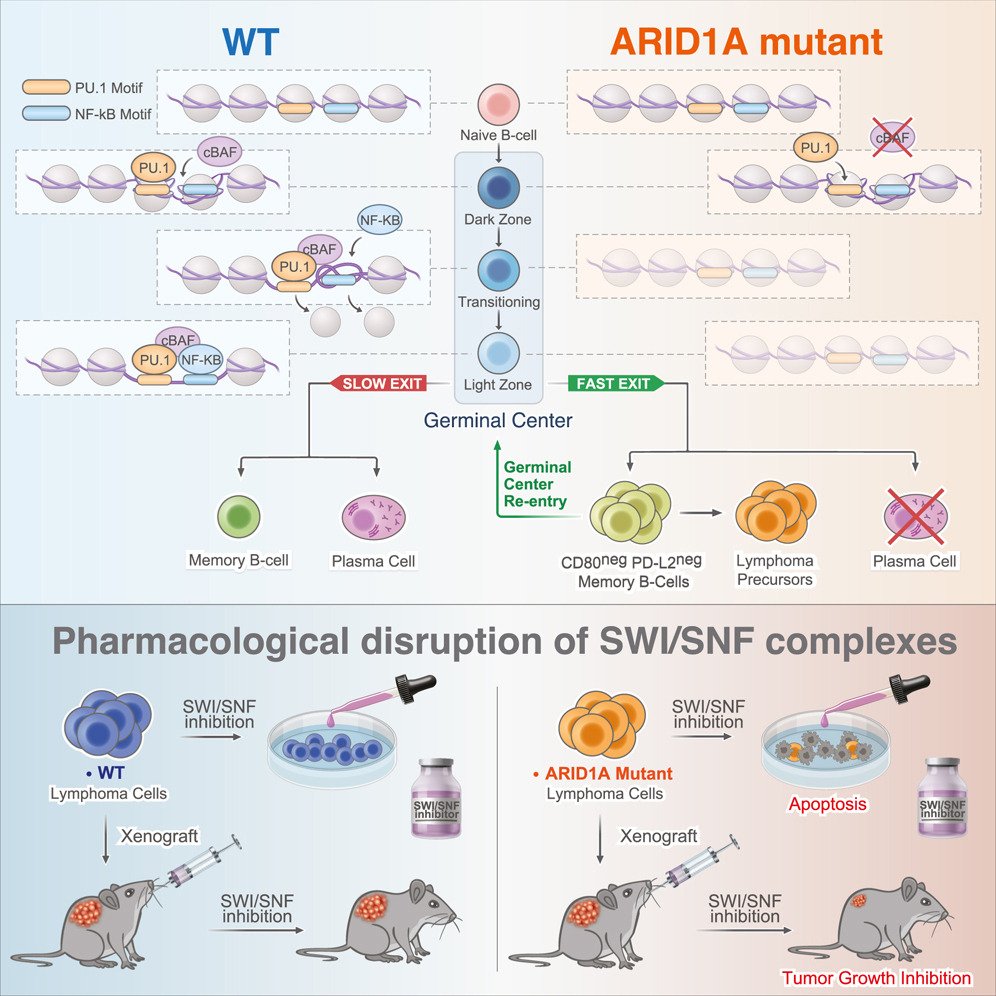
ARID1A orchestrates SWI/SNF-mediated sequential binding of transcription factors with ARID1A loss driving pre-memory B cell fate and lymphomagenesis.
ARID1A, a subunit of the canonical BAF nucleosome remodeling complex, is commonly mutated in lymphomas. We show that ARID1A orchestrates B cell fate during the germinal center (GC) response, facilitating cooperative and sequential binding of PU.1 and NF-kB at crucial genes for cytokine and CD40 signaling. The absence of ARID1A tilts GC cell fate toward immature IgM+CD80−PD-L2− memory B cells, known for their potential to re-enter new GCs. When combined with BCL2 oncogene, ARID1A haploinsufficiency hastens the progression of aggressive follicular lymphomas (FLs) in mice. Patients with FL with ARID1A-inactivating mutations preferentially display an immature memory B cell-like state with increased transformation risk to aggressive disease. These observations offer mechanistic understanding into the emergence of both indolent and aggressive ARID1A-mutant lymphomas through the formation of immature memory-like clonal precursors. Lastly, we demonstrate that ARID1A mutation induces synthetic lethality to SMARCA2/4 inhibition, paving the way for potential precision therapy for high-risk patients.
Journal: Cancer Cell PMID: N/A DOI: 10.1016/j.ccell.2024.02.010

An Aged/Autoimmune B-cell Program Defines the Early Transformation of Extranodal Lymphomas
A third of patients with diffuse large B-cell lymphoma (DLBCL) present with extranodal dissemination, which is associated with inferior clinical outcomes. MYD88L265P is a hallmark extranodal DLBCL mutation that supports lymphoma proliferation. Yet extranodal lymphomagenesis and the role of MYD88L265P in transformation remain mostly unknown. Here, we show that B cells expressing Myd88L252P (MYD88L265P murine equivalent) activate, proliferate, and differentiate with minimal T-cell costimulation. Additionally, Myd88L252P skewed B cells toward memory fate. Unexpectedly, the transcriptional and phenotypic profiles of B cells expressing Myd88L252P, or other extranodal lymphoma founder mutations, resembled those of CD11c+T-BET+ aged/autoimmune memory B cells (AiBC). AiBC-like cells progressively accumulated in animals prone to develop lymphomas, and ablation of T-BET, the AiBC master regulator, stripped mouse and human mutant B cells of their competitive fitness. By identifying a phenotypically defined prospective lymphoma precursor population and its dependencies, our findings pave the way for the early detection of premalignant states and targeted prophylactic interventions in high-risk patients.
Significance:
Extranodal lymphomas feature a very poor prognosis. The identification of phenotypically distinguishable prospective precursor cells represents a milestone in the pursuit of earlier diagnosis, patient stratification, and prophylactic interventions. Conceptually, we found that extranodal lymphomas and autoimmune disorders harness overlapping pathogenic trajectories, suggesting these B-cell disorders develop and evolve within a spectrum.
Journal: Cancer Discovery PMID: 36264161 DOI: 10.1158/2159-8290.CD-22-0561

BCL10 Mutations Define Distinct Dependencies Guiding Precision Therapy for DLBCL
Activated B cell-like diffuse large B-cell lymphomas (ABC-DLBCL) have unfavorable outcomes and chronic activation of CARD11-BCL10-MALT1 (CBM) signal amplification complexes that form due to polymerization of BCL10 subunits, which is affected by recurrent somatic mutations in ABC-DLBCLs. Herein, we show that BCL10 mutants fall into at least two functionally distinct classes: missense mutations of the BCL10 CARD domain and truncation of its C-terminal tail. Truncating mutations abrogated a motif through which MALT1 inhibits BCL10 polymerization, trapping MALT1 in its activated filament-bound state. CARD missense mutations enhanced BCL10 filament formation, forming glutamine network structures that stabilize BCL10 filaments. Mutant forms of BCL10 were less dependent on upstream CARD11 activation and thus manifested resistance to BTK inhibitors, whereas BCL10 truncating but not CARD mutants were hypersensitive to MALT1 inhibitors. Therefore, BCL10 mutations are potential biomarkers for BTK inhibitor resistance in ABC-DLBCL, and further precision can be achieved by selecting therapy based on specific biochemical effects of distinct mutation classes.
Significance: ABC-DLBCLs feature frequent mutations of signaling mediators that converge on the CBM complex. We use structure-function approaches to reveal that BCL10 mutations fall into two distinct biochemical classes. Both classes confer resistance to BTK inhibitors, whereas BCL10 truncations confer hyperresponsiveness to MALT1 inhibitors, providing a road map for precision therapies in ABC-DLBCLs.
Journal: Cancer Discovery PMID: 35658124 DOI: 10.1158/2159-8290.CD-21-1566

SETD2 Haploinsufficiency Enhances Germinal Center–Associated AICDA Somatic Hypermutation to Drive B-cell Lymphomagenesis
SETD2 is the sole histone methyltransferase responsible for H3K36me3, with roles in splicing, transcription initiation, and DNA damage response. Homozygous disruption of SETD2 yields a tumor suppressor effect in various cancers. However, SETD2 mutation is typically heterozygous in diffuse large B-cell lymphomas. Here we show that heterozygous Setd2 deficiency results in germinal center (GC) hyperplasia and increased competitive fitness, with reduced DNA damage checkpoint activity and apoptosis, resulting in accelerated lymphomagenesis. Impaired DNA damage sensing in Setd2-haploinsufficient germinal center B (GCB) and lymphoma cells associated with increased AICDA-induced somatic hypermutation, complex structural variants, and increased translocations including those activating MYC. DNA damage was selectively increased on the nontemplate strand, and H3K36me3 loss was associated with greater RNAPII processivity and mutational burden, suggesting that SETD2-mediated H3K36me3 is required for proper sensing of cytosine deamination. Hence, Setd2 haploinsufficiency delineates a novel GCB context-specific oncogenic pathway involving defective epigenetic surveillance of AICDA-mediated effects on transcribed genes.
Significance: Our findings define a B cell-specific oncogenic effect of SETD2 heterozygous mutation, which unleashes AICDA mutagenesis of nontemplate strand DNA in the GC reaction, resulting in lymphomas with heavy mutational burden. GC-derived lymphomas did not tolerate SETD2 homozygous deletion, pointing to a novel context-specific therapeutic vulnerability.
Journal: Cancer Discovery PMID: 35443279 DOI: 10.1158/2159-8290.CD-21-1514
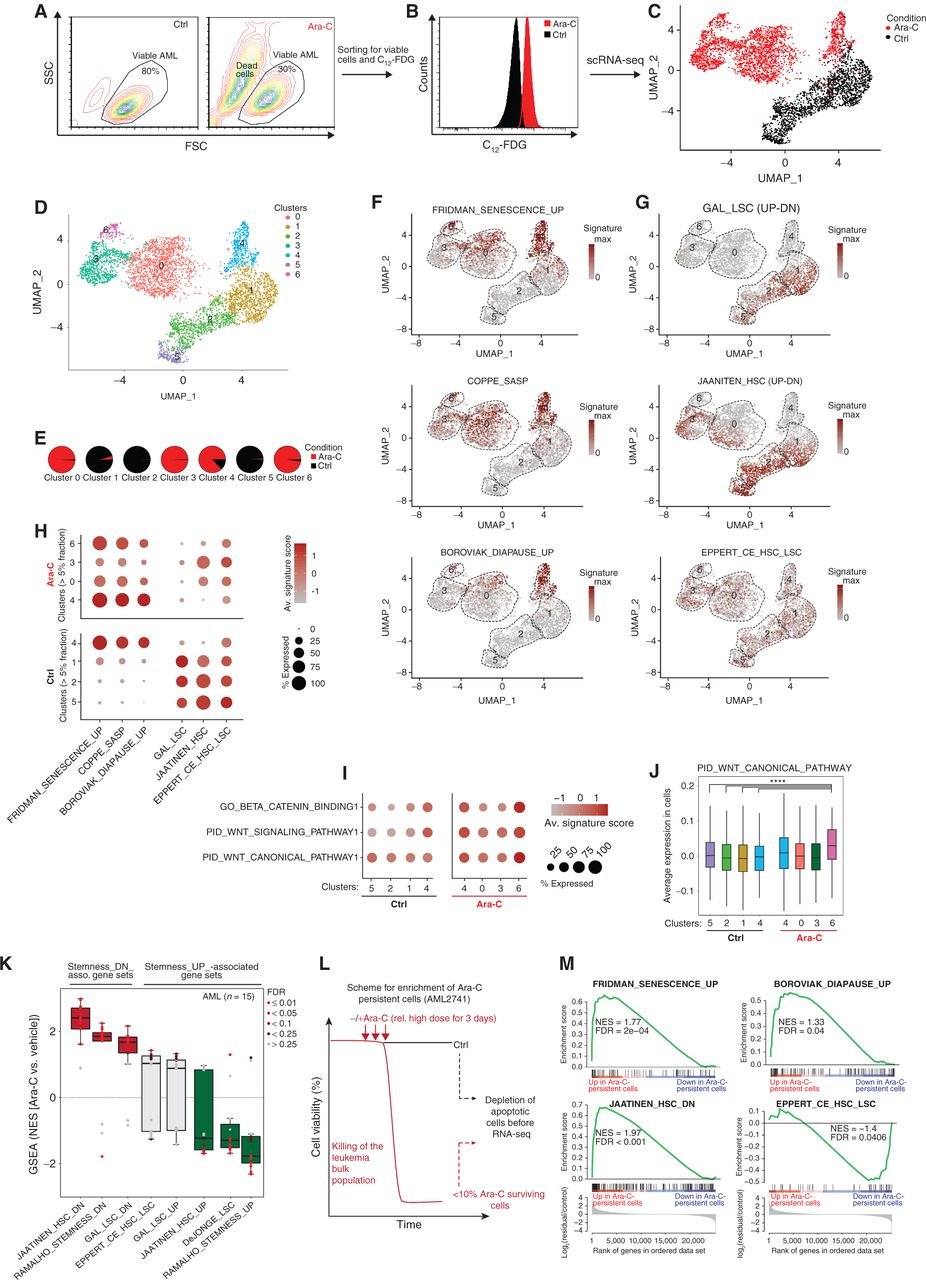
Chemotherapy Induces Senescence-Like Resilient Cells Capable of Initiating AML Recurrence
Patients with acute myeloid leukemia (AML) frequently relapse after chemotherapy, yet the mechanism by which AML reemerges is not fully understood. Herein, we show that primary AML cells enter a senescence-like phenotype following chemotherapy in vitro and in vivo. This is accompanied by induction of senescence/inflammatory and embryonic diapause transcriptional programs, with downregulation of MYC and leukemia stem cell genes. Single-cell RNA sequencing suggested depletion of leukemia stem cells in vitro and in vivo, and enrichment for subpopulations with distinct senescence-like cells. This senescence effect was transient and conferred superior colony-forming and engraftment potential. Entry into this senescence-like phenotype was dependent on ATR, and persistence of AML cells was severely impaired by ATR inhibitors. Altogether, we propose that AML relapse is facilitated by a senescence-like resilience phenotype that occurs regardless of their stem cell status. Upon recovery, these post-senescence AML cells give rise to relapsed AMLs with increased stem cell potential. SIGNIFICANCE: Despite entering complete remission after chemotherapy, relapse occurs in many patients with AML. Thus, there is an urgent need to understand the relapse mechanism in AML and the development of targeted treatments to improve outcome. Here, we identified a senescence-like resilience phenotype through which AML cells can survive and repopulate leukemia.
Journal: Cancer Discovery PMID: 33500244 DOI: 10.1158/2159-8290.CD-20-1375

Smc3 dosage regulates B cell transit through germinal centers and restricts their malignant transformation
During the germinal center (GC) reaction, B cells undergo extensive redistribution of cohesin complex and three-dimensional reorganization of their genomes. Yet, the significance of cohesin and architectural programming in the humoral immune response is unknown. Herein we report that homozygous deletion of Smc3, encoding the cohesin ATPase subunit, abrogated GC formation, while, in marked contrast, Smc3 haploinsufficiency resulted in GC hyperplasia, skewing of GC polarity and impaired plasma cell (PC) differentiation. Genome-wide chromosomal conformation and transcriptional profiling revealed defects in GC B cell terminal differentiation programs controlled by the lymphoma epigenetic tumor suppressors Tet2 and Kmt2d and failure of Smc3-haploinsufficient GC B cells to switch from B cell- to PC-defining transcription factors. Smc3 haploinsufficiency preferentially impaired the connectivity of enhancer elements controlling various lymphoma tumor suppressor genes, and, accordingly, Smc3 haploinsufficiency accelerated lymphomagenesis in mice with constitutive Bcl6 expression. Collectively, our data indicate a dose-dependent function for cohesin in humoral immunity to facilitate the B cell to PC phenotypic switch while restricting malignant transformation.
Journal: Nature Immunology PMID: 33432228 DOI: 10.1038/s41590-020-00827-8

Somatic Mutations Drive Specific, but Reversible, Epigenetic Heterogeneity States in AML
Epigenetic allele diversity is linked to inferior prognosis in acute myeloid leukemia (AML). However, the source of epiallele heterogeneity in AML is unknown. Herein we analyzed epiallele diversity in a genetically and clinically annotated AML cohort. Notably, AML driver mutations linked to transcription factors and favorable outcome are associated with epigenetic destabilization in a defined set of susceptible loci. In contrast, AML subtypes linked to inferior prognosis manifest greater abundance and highly stochastic epiallele patterning. We report an epiallele outcome classifier supporting the link between epigenetic diversity and treatment failure. Mouse models with TET2 or IDH2 mutations show that epiallele diversity is especially strongly induced by IDH mutations, precedes transformation to AML, and is enhanced by cooperation between somatic mutations. Furthermore, epiallele complexity was partially reversed by epigenetic therapies in AML driven by TET2/IDH2, suggesting that epigenetic therapy might function in part by reducing population complexity and fitness of AMLs.
Journal: Cancer Discovery PMID: 32938585 DOI: 10.1158/2159-8290.CD-19-0897

Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response
Follicular lymphomas (FLs) are slow-growing, indolent tumors containing extensive follicular dendritic cell (FDC) networks and recurrent EZH2 gain-of-function mutations. Paradoxically, FLs originate from highly proliferative germinal center (GC) B cells with proliferation strictly dependent on interactions with T follicular helper cells. Herein, we show that EZH2 mutations initiate FL by attenuating GC B cell requirement for T cell help and driving slow expansion of GC centrocytes that become enmeshed with and dependent on FDCs. By impairing T cell help, mutant EZH2 prevents induction of proliferative MYC programs. Thus, EZH2 mutation fosters malignant transformation by epigenetically reprograming B cells to form an aberrant immunological niche that reflects characteristic features of human FLs, explaining how indolent tumors arise from GC B cells.
Journal: Cancer Cell PMID: 32396861 DOI: 10.1016/j.ccell.2020.04.004

Rational Targeting of Cooperating Layers of the Epigenome Yields Enhanced Therapeutic Efficacy against AML
Disruption of epigenetic regulation is a hallmark of acute myeloid leukemia (AML), but epigenetic therapy is complicated by the complexity of the epigenome. Herein, we developed a long-term primary AML ex vivo platform to determine whether targeting different epigenetic layers with 5-azacytidine and LSD1 inhibitors would yield improved efficacy. This combination was most effective in TET2mut AML, where it extinguished leukemia stem cells and particularly induced genes with both LSD1-bound enhancers and cytosine-methylated promoters. Functional studies indicated that derepression of genes such as GATA2 contributes to drug efficacy. Mechanistically, combination therapy increased enhancer–promoter looping and chromatin-activating marks at the GATA2 locus. CRISPRi of the LSD1-bound enhancer in patient-derived TET2mut AML was associated with dampening of therapeutic GATA2 induction. TET2 knockdown in human hematopoietic stem/progenitor cells induced loss of enhancer 5-hydroxymethylation and facilitated LSD1-mediated enhancer inactivation. Our data provide a basis for rational targeting of cooperating aberrant promoter and enhancer epigenetic marks driven by mutant epigenetic modifiers.
Journal: Cancer Discovery PMID: 31076479 DOI: 10.1158/2159-8290.CD-19-0106