
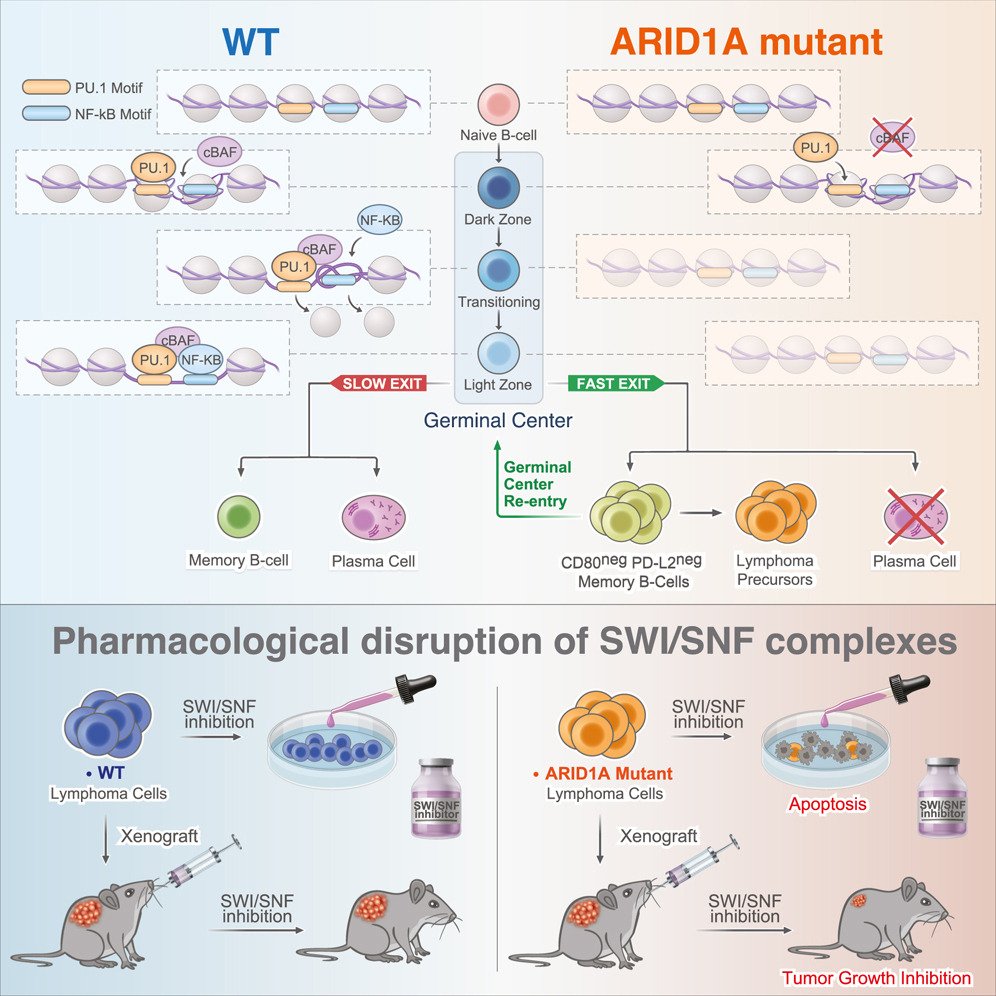
ARID1A orchestrates SWI/SNF-mediated sequential binding of transcription factors with ARID1A loss driving pre-memory B cell fate and lymphomagenesis.
ARID1A, a subunit of the canonical BAF nucleosome remodeling complex, is commonly mutated in lymphomas. We show that ARID1A orchestrates B cell fate during the germinal center (GC) response, facilitating cooperative and sequential binding of PU.1 and NF-kB at crucial genes for cytokine and CD40 signaling. The absence of ARID1A tilts GC cell fate toward immature IgM+CD80−PD-L2− memory B cells, known for their potential to re-enter new GCs. When combined with BCL2 oncogene, ARID1A haploinsufficiency hastens the progression of aggressive follicular lymphomas (FLs) in mice. Patients with FL with ARID1A-inactivating mutations preferentially display an immature memory B cell-like state with increased transformation risk to aggressive disease. These observations offer mechanistic understanding into the emergence of both indolent and aggressive ARID1A-mutant lymphomas through the formation of immature memory-like clonal precursors. Lastly, we demonstrate that ARID1A mutation induces synthetic lethality to SMARCA2/4 inhibition, paving the way for potential precision therapy for high-risk patients.
Journal: Cancer Cell PMID: N/A DOI: 10.1016/j.ccell.2024.02.010

SETD2 Haploinsufficiency Enhances Germinal Center–Associated AICDA Somatic Hypermutation to Drive B-cell Lymphomagenesis
SETD2 is the sole histone methyltransferase responsible for H3K36me3, with roles in splicing, transcription initiation, and DNA damage response. Homozygous disruption of SETD2 yields a tumor suppressor effect in various cancers. However, SETD2 mutation is typically heterozygous in diffuse large B-cell lymphomas. Here we show that heterozygous Setd2 deficiency results in germinal center (GC) hyperplasia and increased competitive fitness, with reduced DNA damage checkpoint activity and apoptosis, resulting in accelerated lymphomagenesis. Impaired DNA damage sensing in Setd2-haploinsufficient germinal center B (GCB) and lymphoma cells associated with increased AICDA-induced somatic hypermutation, complex structural variants, and increased translocations including those activating MYC. DNA damage was selectively increased on the nontemplate strand, and H3K36me3 loss was associated with greater RNAPII processivity and mutational burden, suggesting that SETD2-mediated H3K36me3 is required for proper sensing of cytosine deamination. Hence, Setd2 haploinsufficiency delineates a novel GCB context-specific oncogenic pathway involving defective epigenetic surveillance of AICDA-mediated effects on transcribed genes.
Significance: Our findings define a B cell-specific oncogenic effect of SETD2 heterozygous mutation, which unleashes AICDA mutagenesis of nontemplate strand DNA in the GC reaction, resulting in lymphomas with heavy mutational burden. GC-derived lymphomas did not tolerate SETD2 homozygous deletion, pointing to a novel context-specific therapeutic vulnerability.
Journal: Cancer Discovery PMID: 35443279 DOI: 10.1158/2159-8290.CD-21-1514
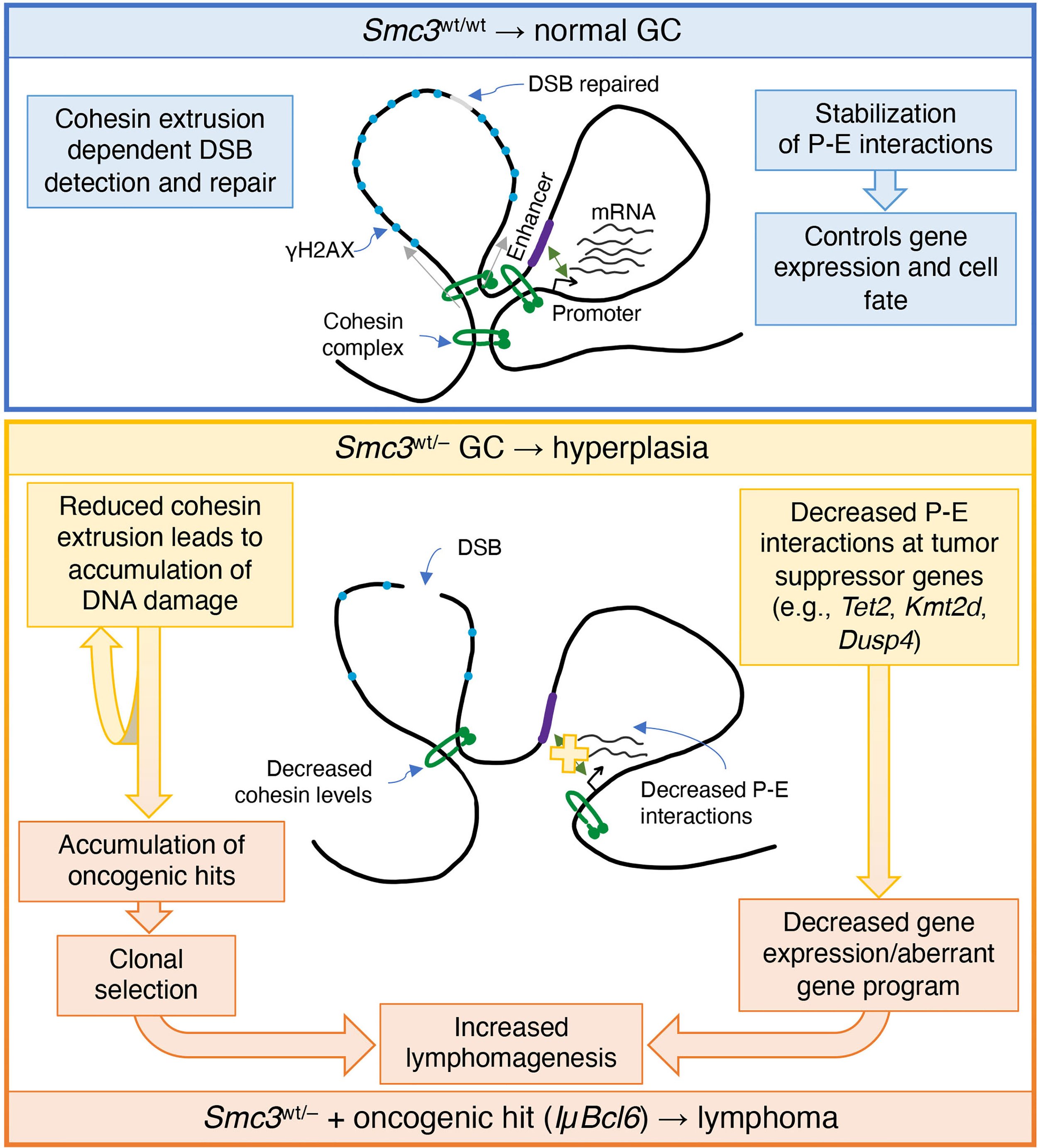
Cohesin Core Complex Gene Dosage Contributes to Germinal Center Derived Lymphoma Phenotypes and Outcomes
The cohesin complex plays critical roles in genomic stability and gene expression through effects on 3D architecture. Cohesin core subunit genes are mutated across a wide cross-section of cancers, but not in germinal center (GC) derived lymphomas. In spite of this, haploinsufficiency of cohesin ATPase subunit Smc3 was shown to contribute to malignant transformation of GC B-cells in mice. Herein we explored potential mechanisms and clinical relevance of Smc3 deficiency in GC lymphomagenesis. Transcriptional profiling of Smc3 haploinsufficient murine lymphomas revealed downregulation of genes repressed by loss of epigenetic tumor suppressors Tet2 and Kmt2d. Profiling 3D chromosomal interactions in lymphomas revealed impaired enhancer-promoter interactions affecting genes like Tet2, which was aberrantly downregulated in Smc3 deficient lymphomas. Tet2 plays important roles in B-cell exit from the GC reaction, and single cell RNA-seq profiles and phenotypic trajectory analysis in Smc3 mutant mice revealed a specific defect in commitment to the final steps of plasma cell differentiation. Although Smc3 deficiency resulted in structural abnormalities in GC B-cells, there was no increase of somatic mutations or structural variants in Smc3 haploinsufficient lymphomas, suggesting that cohesin deficiency largely induces lymphomas through disruption of enhancer-promoter interactions of terminal differentiation and tumor suppressor genes. Strikingly, the presence of the Smc3 haploinsufficient GC B-cell transcriptional signature in human patients with GC-derived diffuse large B-cell lymphoma (DLBCL) was linked to inferior clinical outcome and low expression of cohesin core subunits. Reciprocally, reduced expression of cohesin subunits was an independent risk factor for worse survival int DLBCL patient cohorts. Collectively, the data suggest that Smc3 functions as a bona fide tumor suppressor for lymphomas through non-genetic mechanisms, and drives disease by disrupting the commitment of GC B-cells to the plasma cell fate.
Journal: Frontiers Immunology PMID: 34621263 DOI: 10.3389/fimmu.2021.688493
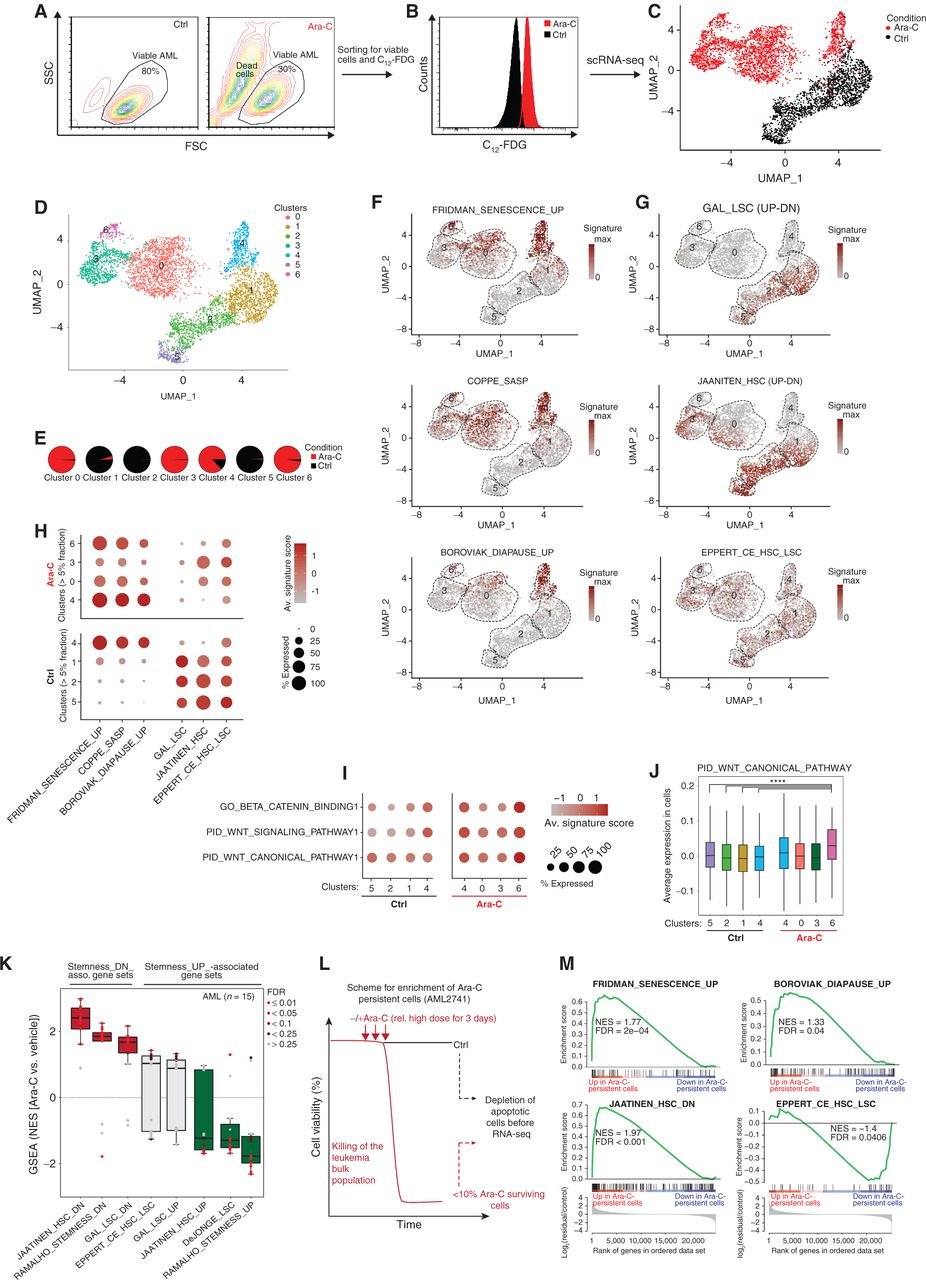
Chemotherapy Induces Senescence-Like Resilient Cells Capable of Initiating AML Recurrence
Patients with acute myeloid leukemia (AML) frequently relapse after chemotherapy, yet the mechanism by which AML reemerges is not fully understood. Herein, we show that primary AML cells enter a senescence-like phenotype following chemotherapy in vitro and in vivo. This is accompanied by induction of senescence/inflammatory and embryonic diapause transcriptional programs, with downregulation of MYC and leukemia stem cell genes. Single-cell RNA sequencing suggested depletion of leukemia stem cells in vitro and in vivo, and enrichment for subpopulations with distinct senescence-like cells. This senescence effect was transient and conferred superior colony-forming and engraftment potential. Entry into this senescence-like phenotype was dependent on ATR, and persistence of AML cells was severely impaired by ATR inhibitors. Altogether, we propose that AML relapse is facilitated by a senescence-like resilience phenotype that occurs regardless of their stem cell status. Upon recovery, these post-senescence AML cells give rise to relapsed AMLs with increased stem cell potential. SIGNIFICANCE: Despite entering complete remission after chemotherapy, relapse occurs in many patients with AML. Thus, there is an urgent need to understand the relapse mechanism in AML and the development of targeted treatments to improve outcome. Here, we identified a senescence-like resilience phenotype through which AML cells can survive and repopulate leukemia.
Journal: Cancer Discovery PMID: 33500244 DOI: 10.1158/2159-8290.CD-20-1375