
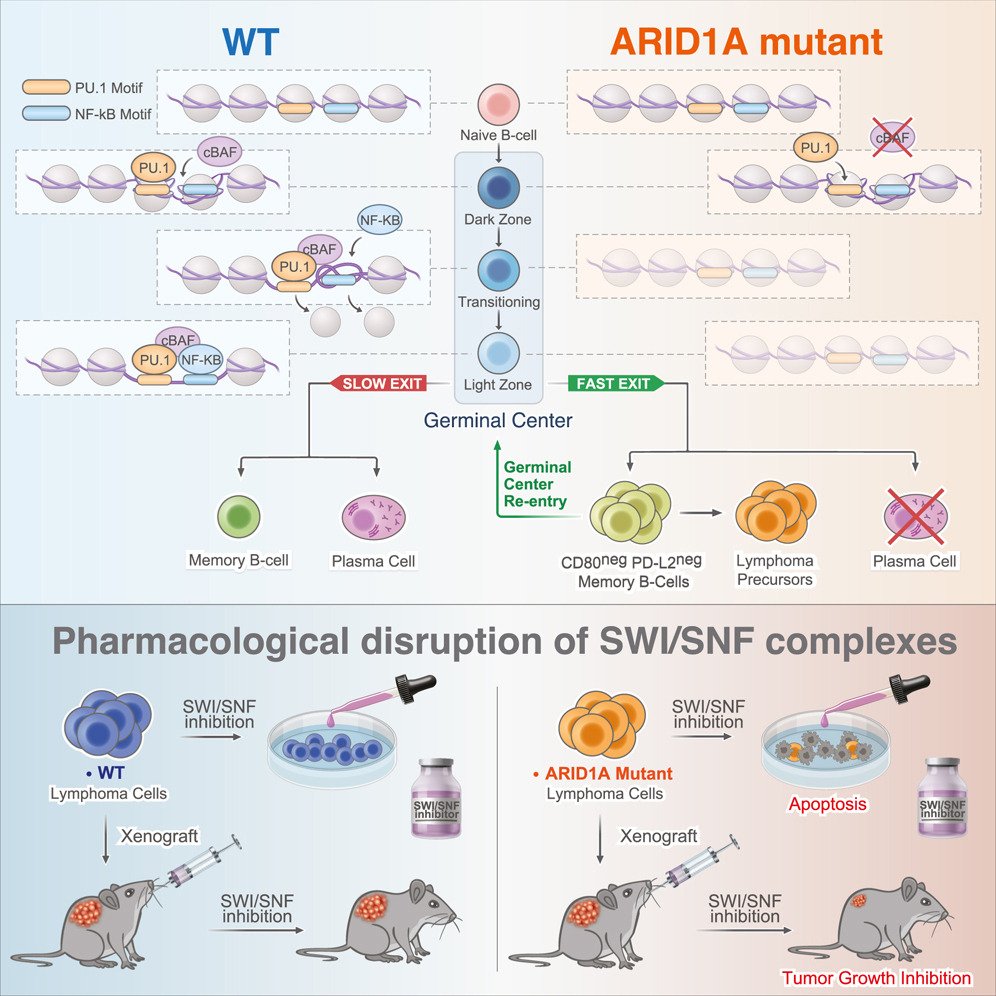
ARID1A orchestrates SWI/SNF-mediated sequential binding of transcription factors with ARID1A loss driving pre-memory B cell fate and lymphomagenesis.
ARID1A, a subunit of the canonical BAF nucleosome remodeling complex, is commonly mutated in lymphomas. We show that ARID1A orchestrates B cell fate during the germinal center (GC) response, facilitating cooperative and sequential binding of PU.1 and NF-kB at crucial genes for cytokine and CD40 signaling. The absence of ARID1A tilts GC cell fate toward immature IgM+CD80−PD-L2− memory B cells, known for their potential to re-enter new GCs. When combined with BCL2 oncogene, ARID1A haploinsufficiency hastens the progression of aggressive follicular lymphomas (FLs) in mice. Patients with FL with ARID1A-inactivating mutations preferentially display an immature memory B cell-like state with increased transformation risk to aggressive disease. These observations offer mechanistic understanding into the emergence of both indolent and aggressive ARID1A-mutant lymphomas through the formation of immature memory-like clonal precursors. Lastly, we demonstrate that ARID1A mutation induces synthetic lethality to SMARCA2/4 inhibition, paving the way for potential precision therapy for high-risk patients.
Journal: Cancer Cell PMID: N/A DOI: 10.1016/j.ccell.2024.02.010

An Aged/Autoimmune B-cell Program Defines the Early Transformation of Extranodal Lymphomas
A third of patients with diffuse large B-cell lymphoma (DLBCL) present with extranodal dissemination, which is associated with inferior clinical outcomes. MYD88L265P is a hallmark extranodal DLBCL mutation that supports lymphoma proliferation. Yet extranodal lymphomagenesis and the role of MYD88L265P in transformation remain mostly unknown. Here, we show that B cells expressing Myd88L252P (MYD88L265P murine equivalent) activate, proliferate, and differentiate with minimal T-cell costimulation. Additionally, Myd88L252P skewed B cells toward memory fate. Unexpectedly, the transcriptional and phenotypic profiles of B cells expressing Myd88L252P, or other extranodal lymphoma founder mutations, resembled those of CD11c+T-BET+ aged/autoimmune memory B cells (AiBC). AiBC-like cells progressively accumulated in animals prone to develop lymphomas, and ablation of T-BET, the AiBC master regulator, stripped mouse and human mutant B cells of their competitive fitness. By identifying a phenotypically defined prospective lymphoma precursor population and its dependencies, our findings pave the way for the early detection of premalignant states and targeted prophylactic interventions in high-risk patients.
Significance:
Extranodal lymphomas feature a very poor prognosis. The identification of phenotypically distinguishable prospective precursor cells represents a milestone in the pursuit of earlier diagnosis, patient stratification, and prophylactic interventions. Conceptually, we found that extranodal lymphomas and autoimmune disorders harness overlapping pathogenic trajectories, suggesting these B-cell disorders develop and evolve within a spectrum.
Journal: Cancer Discovery PMID: 36264161 DOI: 10.1158/2159-8290.CD-22-0561

BCL10 Mutations Define Distinct Dependencies Guiding Precision Therapy for DLBCL
Activated B cell-like diffuse large B-cell lymphomas (ABC-DLBCL) have unfavorable outcomes and chronic activation of CARD11-BCL10-MALT1 (CBM) signal amplification complexes that form due to polymerization of BCL10 subunits, which is affected by recurrent somatic mutations in ABC-DLBCLs. Herein, we show that BCL10 mutants fall into at least two functionally distinct classes: missense mutations of the BCL10 CARD domain and truncation of its C-terminal tail. Truncating mutations abrogated a motif through which MALT1 inhibits BCL10 polymerization, trapping MALT1 in its activated filament-bound state. CARD missense mutations enhanced BCL10 filament formation, forming glutamine network structures that stabilize BCL10 filaments. Mutant forms of BCL10 were less dependent on upstream CARD11 activation and thus manifested resistance to BTK inhibitors, whereas BCL10 truncating but not CARD mutants were hypersensitive to MALT1 inhibitors. Therefore, BCL10 mutations are potential biomarkers for BTK inhibitor resistance in ABC-DLBCL, and further precision can be achieved by selecting therapy based on specific biochemical effects of distinct mutation classes.
Significance: ABC-DLBCLs feature frequent mutations of signaling mediators that converge on the CBM complex. We use structure-function approaches to reveal that BCL10 mutations fall into two distinct biochemical classes. Both classes confer resistance to BTK inhibitors, whereas BCL10 truncations confer hyperresponsiveness to MALT1 inhibitors, providing a road map for precision therapies in ABC-DLBCLs.
Journal: Cancer Discovery PMID: 35658124 DOI: 10.1158/2159-8290.CD-21-1566