
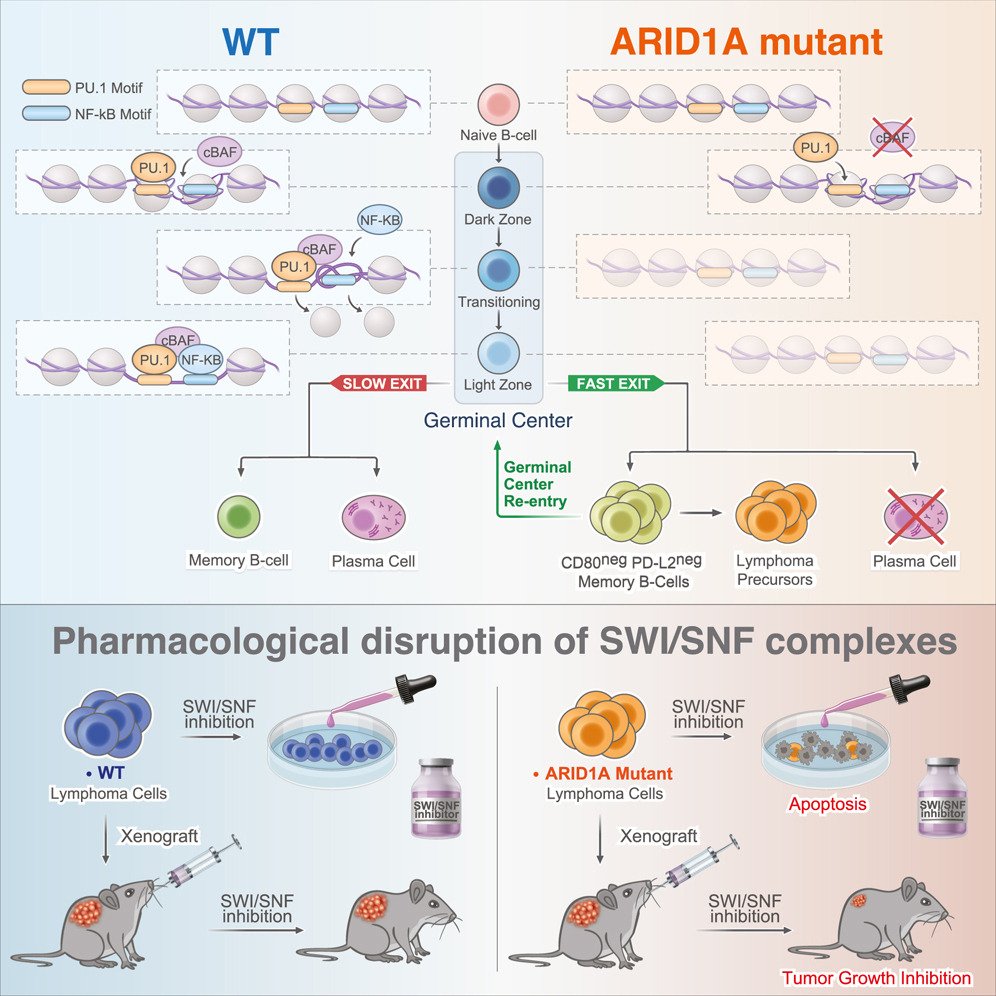
ARID1A orchestrates SWI/SNF-mediated sequential binding of transcription factors with ARID1A loss driving pre-memory B cell fate and lymphomagenesis.
ARID1A, a subunit of the canonical BAF nucleosome remodeling complex, is commonly mutated in lymphomas. We show that ARID1A orchestrates B cell fate during the germinal center (GC) response, facilitating cooperative and sequential binding of PU.1 and NF-kB at crucial genes for cytokine and CD40 signaling. The absence of ARID1A tilts GC cell fate toward immature IgM+CD80−PD-L2− memory B cells, known for their potential to re-enter new GCs. When combined with BCL2 oncogene, ARID1A haploinsufficiency hastens the progression of aggressive follicular lymphomas (FLs) in mice. Patients with FL with ARID1A-inactivating mutations preferentially display an immature memory B cell-like state with increased transformation risk to aggressive disease. These observations offer mechanistic understanding into the emergence of both indolent and aggressive ARID1A-mutant lymphomas through the formation of immature memory-like clonal precursors. Lastly, we demonstrate that ARID1A mutation induces synthetic lethality to SMARCA2/4 inhibition, paving the way for potential precision therapy for high-risk patients.
Journal: Cancer Cell PMID: N/A DOI: 10.1016/j.ccell.2024.02.010

BTG1 mutation yields supercompetitive B cells primed for malignant transformation
Multicellular life requires altruistic cooperation between cells. The adaptive immune system is a notable exception, wherein germinal center B cells compete vigorously for limiting positive selection signals. Studying primary human lymphomas and developing new mouse models, we found that mutations affecting BTG1 disrupt a critical immune gatekeeper mechanism that strictly limits B cell fitness during antibody affinity maturation. This mechanism converted germinal center B cells into supercompetitors that rapidly outstrip their normal counterparts. This effect was conferred by a small shift in MYC protein induction kinetics but resulted in aggressive invasive lymphomas, which in humans are linked to dire clinical outcomes. Our findings reveal a delicate evolutionary trade-off between natural selection of B cells to provide immunity and potentially dangerous features that recall the more competitive nature of unicellular organisms.
Journal: Science PMID: 36656933 DOI: 10.1126/science.abj7412

SETD2 Haploinsufficiency Enhances Germinal Center–Associated AICDA Somatic Hypermutation to Drive B-cell Lymphomagenesis
SETD2 is the sole histone methyltransferase responsible for H3K36me3, with roles in splicing, transcription initiation, and DNA damage response. Homozygous disruption of SETD2 yields a tumor suppressor effect in various cancers. However, SETD2 mutation is typically heterozygous in diffuse large B-cell lymphomas. Here we show that heterozygous Setd2 deficiency results in germinal center (GC) hyperplasia and increased competitive fitness, with reduced DNA damage checkpoint activity and apoptosis, resulting in accelerated lymphomagenesis. Impaired DNA damage sensing in Setd2-haploinsufficient germinal center B (GCB) and lymphoma cells associated with increased AICDA-induced somatic hypermutation, complex structural variants, and increased translocations including those activating MYC. DNA damage was selectively increased on the nontemplate strand, and H3K36me3 loss was associated with greater RNAPII processivity and mutational burden, suggesting that SETD2-mediated H3K36me3 is required for proper sensing of cytosine deamination. Hence, Setd2 haploinsufficiency delineates a novel GCB context-specific oncogenic pathway involving defective epigenetic surveillance of AICDA-mediated effects on transcribed genes.
Significance: Our findings define a B cell-specific oncogenic effect of SETD2 heterozygous mutation, which unleashes AICDA mutagenesis of nontemplate strand DNA in the GC reaction, resulting in lymphomas with heavy mutational burden. GC-derived lymphomas did not tolerate SETD2 homozygous deletion, pointing to a novel context-specific therapeutic vulnerability.
Journal: Cancer Discovery PMID: 35443279 DOI: 10.1158/2159-8290.CD-21-1514

TBL1XR1 Mutations Drive Extranodal Lymphoma by Inducing a Pro-tumorigenic Memory Fate
The most aggressive B cell lymphomas frequently manifest extranodal distribution and carry somatic mutations in the poorly characterized gene TBL1XR1. Here, we show that TBL1XR1 mutations skew the humoral immune response toward generating abnormal immature memory B cells (MB), while impairing plasma cell differentiation. At the molecular level, TBL1XR1 mutants co-opt SMRT/HDAC3 repressor complexes toward binding the MB cell transcription factor (TF) BACH2 at the expense of the germinal center (GC) TF BCL6, leading to pre-memory transcriptional reprogramming and cell-fate bias. Upon antigen recall, TBL1XR1 mutant MB cells fail to differentiate into plasma cells and instead preferentially reenter new GC reactions, providing evidence for a cyclic reentry lymphomagenesis mechanism. Ultimately, TBL1XR1 alterations lead to a striking extranodal immunoblastic lymphoma phenotype that mimics the human disease. Both human and murine lymphomas feature expanded MB-like cell populations, consistent with a MB-cell origin and delineating an unforeseen pathway for malignant transformation of the immune system.
Journal: Cell PMID: 32619424 DOI: 10.1016/j.cell.2020.05.049