
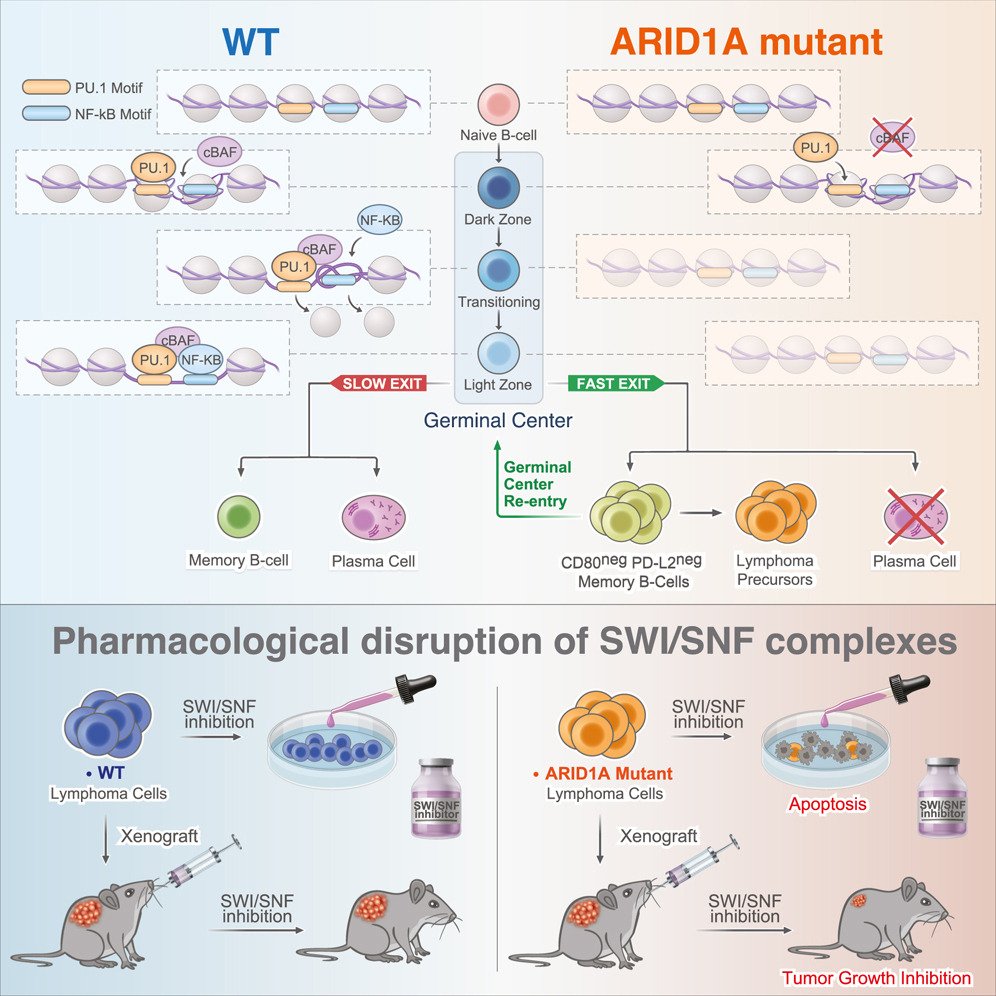
ARID1A orchestrates SWI/SNF-mediated sequential binding of transcription factors with ARID1A loss driving pre-memory B cell fate and lymphomagenesis.
ARID1A, a subunit of the canonical BAF nucleosome remodeling complex, is commonly mutated in lymphomas. We show that ARID1A orchestrates B cell fate during the germinal center (GC) response, facilitating cooperative and sequential binding of PU.1 and NF-kB at crucial genes for cytokine and CD40 signaling. The absence of ARID1A tilts GC cell fate toward immature IgM+CD80−PD-L2− memory B cells, known for their potential to re-enter new GCs. When combined with BCL2 oncogene, ARID1A haploinsufficiency hastens the progression of aggressive follicular lymphomas (FLs) in mice. Patients with FL with ARID1A-inactivating mutations preferentially display an immature memory B cell-like state with increased transformation risk to aggressive disease. These observations offer mechanistic understanding into the emergence of both indolent and aggressive ARID1A-mutant lymphomas through the formation of immature memory-like clonal precursors. Lastly, we demonstrate that ARID1A mutation induces synthetic lethality to SMARCA2/4 inhibition, paving the way for potential precision therapy for high-risk patients.
Journal: Cancer Cell PMID: N/A DOI: 10.1016/j.ccell.2024.02.010

An Aged/Autoimmune B-cell Program Defines the Early Transformation of Extranodal Lymphomas
A third of patients with diffuse large B-cell lymphoma (DLBCL) present with extranodal dissemination, which is associated with inferior clinical outcomes. MYD88L265P is a hallmark extranodal DLBCL mutation that supports lymphoma proliferation. Yet extranodal lymphomagenesis and the role of MYD88L265P in transformation remain mostly unknown. Here, we show that B cells expressing Myd88L252P (MYD88L265P murine equivalent) activate, proliferate, and differentiate with minimal T-cell costimulation. Additionally, Myd88L252P skewed B cells toward memory fate. Unexpectedly, the transcriptional and phenotypic profiles of B cells expressing Myd88L252P, or other extranodal lymphoma founder mutations, resembled those of CD11c+T-BET+ aged/autoimmune memory B cells (AiBC). AiBC-like cells progressively accumulated in animals prone to develop lymphomas, and ablation of T-BET, the AiBC master regulator, stripped mouse and human mutant B cells of their competitive fitness. By identifying a phenotypically defined prospective lymphoma precursor population and its dependencies, our findings pave the way for the early detection of premalignant states and targeted prophylactic interventions in high-risk patients.
Significance:
Extranodal lymphomas feature a very poor prognosis. The identification of phenotypically distinguishable prospective precursor cells represents a milestone in the pursuit of earlier diagnosis, patient stratification, and prophylactic interventions. Conceptually, we found that extranodal lymphomas and autoimmune disorders harness overlapping pathogenic trajectories, suggesting these B-cell disorders develop and evolve within a spectrum.
Journal: Cancer Discovery PMID: 36264161 DOI: 10.1158/2159-8290.CD-22-0561

TBL1XR1 Mutations Drive Extranodal Lymphoma by Inducing a Pro-tumorigenic Memory Fate
The most aggressive B cell lymphomas frequently manifest extranodal distribution and carry somatic mutations in the poorly characterized gene TBL1XR1. Here, we show that TBL1XR1 mutations skew the humoral immune response toward generating abnormal immature memory B cells (MB), while impairing plasma cell differentiation. At the molecular level, TBL1XR1 mutants co-opt SMRT/HDAC3 repressor complexes toward binding the MB cell transcription factor (TF) BACH2 at the expense of the germinal center (GC) TF BCL6, leading to pre-memory transcriptional reprogramming and cell-fate bias. Upon antigen recall, TBL1XR1 mutant MB cells fail to differentiate into plasma cells and instead preferentially reenter new GC reactions, providing evidence for a cyclic reentry lymphomagenesis mechanism. Ultimately, TBL1XR1 alterations lead to a striking extranodal immunoblastic lymphoma phenotype that mimics the human disease. Both human and murine lymphomas feature expanded MB-like cell populations, consistent with a MB-cell origin and delineating an unforeseen pathway for malignant transformation of the immune system.
Journal: Cell PMID: 32619424 DOI: 10.1016/j.cell.2020.05.049

Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response
Follicular lymphomas (FLs) are slow-growing, indolent tumors containing extensive follicular dendritic cell (FDC) networks and recurrent EZH2 gain-of-function mutations. Paradoxically, FLs originate from highly proliferative germinal center (GC) B cells with proliferation strictly dependent on interactions with T follicular helper cells. Herein, we show that EZH2 mutations initiate FL by attenuating GC B cell requirement for T cell help and driving slow expansion of GC centrocytes that become enmeshed with and dependent on FDCs. By impairing T cell help, mutant EZH2 prevents induction of proliferative MYC programs. Thus, EZH2 mutation fosters malignant transformation by epigenetically reprograming B cells to form an aberrant immunological niche that reflects characteristic features of human FLs, explaining how indolent tumors arise from GC B cells.
Journal: Cancer Cell PMID: 32396861 DOI: 10.1016/j.ccell.2020.04.004